
People
Johannes Eller (Alumnus)
Elmar Sauer (Alumnus)
The group of S. Majid Hassanizadeh and Amir Raoof are expected to continue and intensify their scientific work in the framework of SFB 1313. Research Project A01 clarifies thermodynamics phenomena of porous materials in using DFT as a well-founded diffuse interface approach with average molecular resolution. In cooperation with Research Project A02 hydrophilic and hydrophobic materials, but also changes in wettability will be studied. These aspects are also particularly relevant on the small scale and will be studied in cooperation with the group of Majid Hassanizadeh and Amir Raoof at Utrecht University. Joint investigations of free flow/porous-media flow have started.
Publications
(Journal-) Articles
- Bursik, B., Eller, J., & Gross, J. (2024). Predicting Solvation Free Energies from the Minnesota Solvation Database Using Classical Density Functional Theory Based on the PC-SAFT Equation of State. The Journal of Physical Chemistry B, 128, Article 15. https://doi.org/10.1021/acs.jpcb.3c07447
- Stierle, R., Bauer, G., Thiele, N., Bursik, B., Rehner, P., & Gross, J. (2024). Classical density functional theory in three dimensions with GPU-accelerated automatic differentiation: Computational performance analysis using the example of adsorption in covalent-organic frameworks. Chemical Engineering Science, 298, 120380. https://doi.org/10.1016/j.ces.2024.120380
- van Westen, T., Hammer, M., Hafskjold, B., Aasen, A., Gross, J., & Wilhelmsen, Ø. (2022). Perturbation theories for fluids with short-ranged attractive forces: A case study of the Lennard-Jones spline fluid. The Journal of Chemical Physics, 156, Article 10. https://doi.org/10.1063/5.0082690
- Eller, J., Matzerath, T., van Westen, T., & Gross, J. (2021). Predicting solvation free energies in non-polar solvents using classical density functional theory based on the PC-SAFT equation of state. The Journal of Chemical Physics, 154, Article 24. https://doi.org/10.1063/5.0051201
- Stierle, R., & Gross, J. (2021). Hydrodynamic density functional theory for mixtures from a variational principle and its application to droplet coalescence. The Journal of Chemical Physics, 155, Article 13. https://doi.org/10.1063/5.0060088
- Stierle, R., Sauer, E., Eller, J., Theiss, M., Rehner, P., Ackermann, P., & Gross, J. (2020). Guide to efficient solution of PC-SAFT classical Density Functional Theory in various Coordinate Systems using fast Fourier and similar Transforms. Fluid Phase Equilibria, 504, 112306. https://doi.org/10.1016/j.fluid.2019.112306
- Sauer, E., Terzis, A., Theiss, M., Weigand, B., & Gross, J. (2018). Prediction of Contact Angles and Density Profiles of Sessile Droplets Using Classical Density Functional Theory Based on the PCP-SAFT Equation of State. Langmuir, 34, Article 42. https://doi.org/10.1021/acs.langmuir.8b01985
Published data sets
- Bursik, B., Eller, J., & Gross, J. (2024). Supporting Information: Notebooks, Solute Configurations and Solvation Free Energy Data [DaRUS]. https://doi.org/10.18419/DARUS-3756
- Bursik, B., Stierle, R., Schlaich, A., Rehner, P., & Gross, J. (2024). Additional Material: Viscosities of Inhomogeneous Systems from Generalized Entropy Scaling [DaRUS]. https://doi.org/10.18419/DARUS-3769
Research
About this Project
Most porous materials involve non-uniform solid surfaces. This project applies and further develops classical density functional theory (DFT) to study how chemical and structural heterogeneities of the porous material alter adsorption isotherms, wetting behaviour, surface tensions, and contact angles. With these developments, the project allows meaningful predictions of interfacial properties and adorption properties. The equilibrium model serves as a basis for developing a dynamic DFT approach in a forthcoming project period.
Results
The primary objective of this research project is the accurate prediction of Young contact angles and adsorption isotherms of fluids in heterogeneous porous media. Most existing applications for the study of adsorption phenomena using DFT only consider spherical species in 1-dimensional slit shaped pores. Such systems provide a simple way to assess the accuracy of DFT models by comparing the inhomogeneous density distribution inside the pore and the resulting adsorption isotherms to molecular simulations. A comparison to adsorption experiments of real porous material, however, illustrates that the adsorption behaviour of these materials is poorly represented by the 1-dimensional slit pore model due to the inhomogeneous pore structure of porous media, both in pore size distribution and the chemical composition of nanopores.
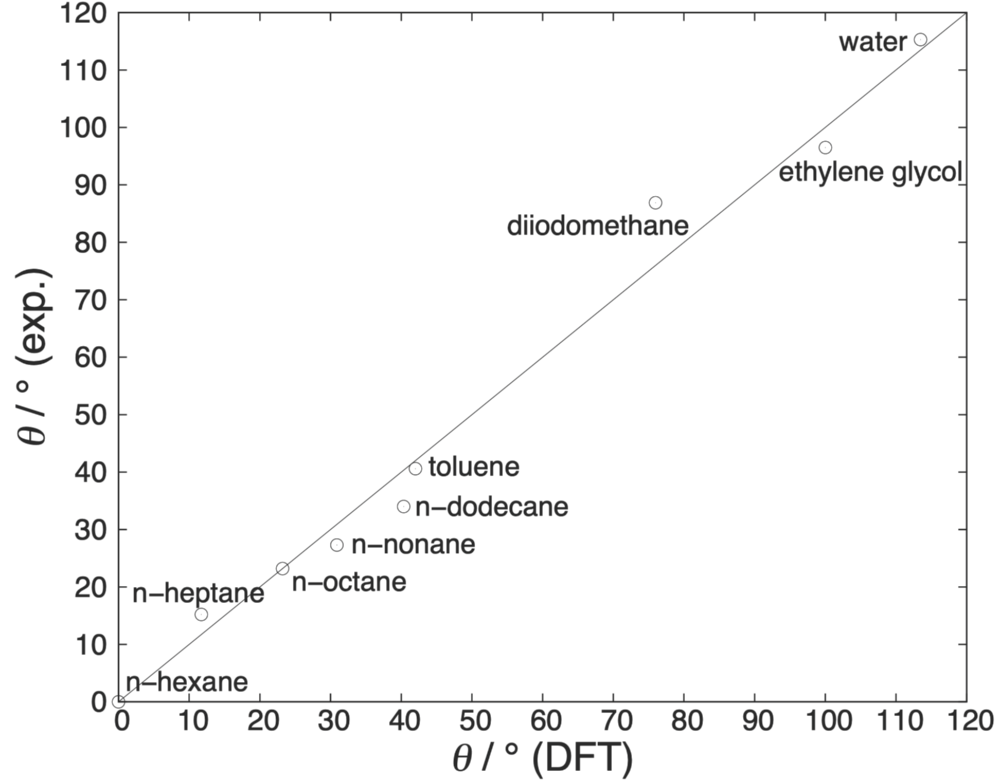
We also regarded sessile droplets and the prediction of Young contact angles, where we compared DFT results to own molecular simulation data and experiments. Here, we calculated the inhomogeneous density profile of sessile ethane droplets with DFT and molecular simulations. The microscopic contact angle can be inferred from a contact line analysis. Macroscopic Young contact angles are calculated from the individual interfacial tensions between solid, fluid and vapor phase. The resulting contact angles can be correlated to experiments where we observe very good agreement even for strongly dipolar fluids like water.
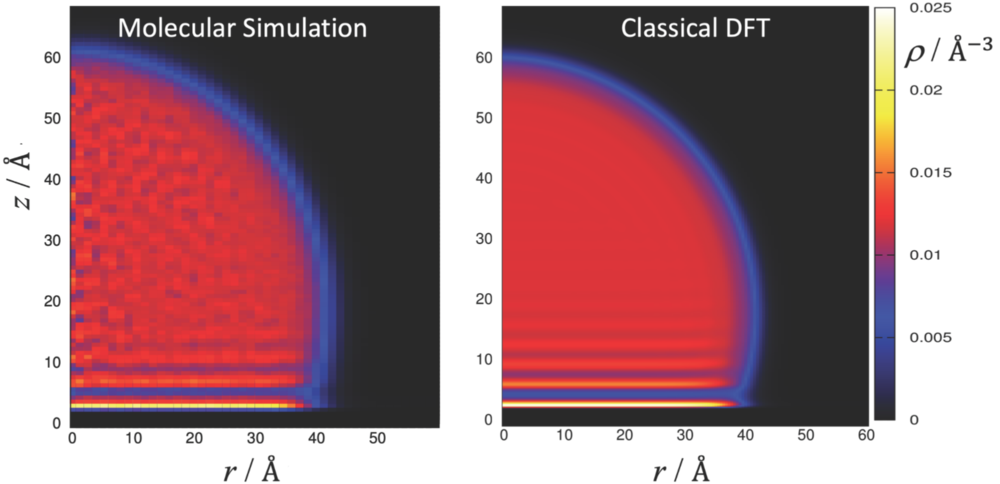
Accounting for chemical heterogeneity and molecular roughness on the nanoscale to better replicate the adsorption behaviour is one of the project goals. We consider 2-dimensional model pores for model development and consider 3-dimensional ordered porous media, like Covalent Organic Frameworks and zeolites, to facilitate a comparison between DFT and own molecular simulation results.
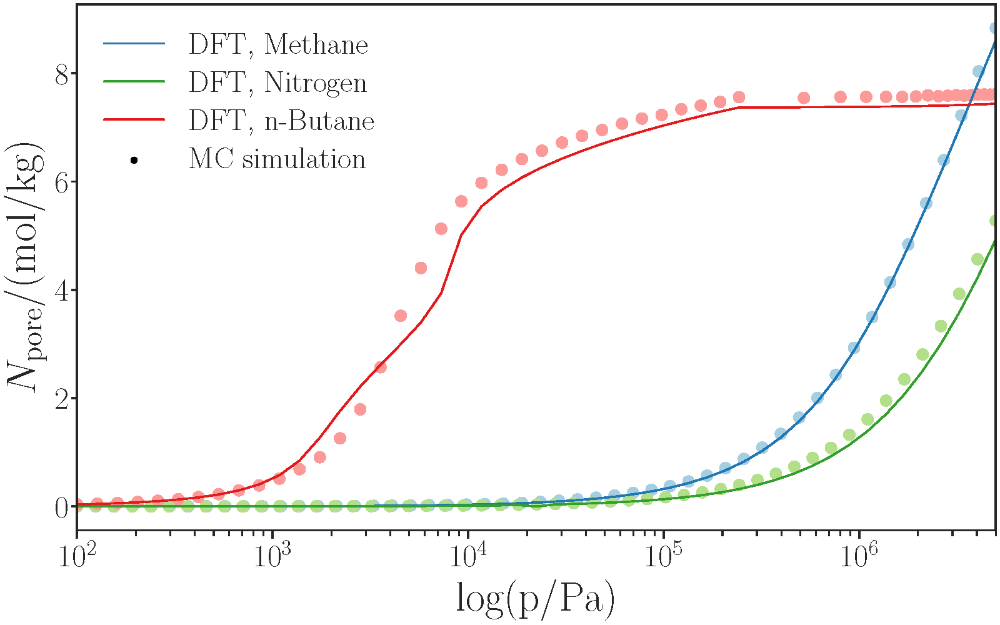
Adsorption isotherms of light gases, like nitrogen, methane and n-butane, in 3-dimensional were predicted in zeolites. Excellent agreement between adsorption isotherms from DFT and molecular simulations are observed without adjustable model parameters, which highlights the strength of the DFT formalism even for highly confined pore geometries.
The significant time savings by DFT can be further amplified by exploiting the cylindral symmetry of the pore channels. For Covalent Organic Frameworks we reduce the dimensionality of the system to 1 dimension by introducing free-energy averaged potentials. This averaged potential contains all information regarding the inhomogeneous pore structure and is able to faithfully represent the adsorption behaviour.

Future Work
Current research focusses on the extension from static properties to modelling time-dependent processes for mixtures by combining classical density functional theory with component and momentum balance equations. This approach allows the modeling of interfacial processes on the nanoscale like contact angle hysteresis, droplet evaporation and coalescence. A central obective is the integration of the hydrodynamic DFT framework into the DuMux simulation toolbox. This enables the study of interfacial phenomena at multiple scales and facilitates the use of DFT models in a broader user-community.
For further information please contact

Joachim Groß
Prof. Dr.-Ing.Project Leader, Research Project A01